Paroxysmale nächtliche Hämoglobinurie: Klinische Merkmale, Laboruntersuchungen und Behandlung
Paroxysmale nächtliche Hämoglobinurie: Klinische Merkmale, Laboruntersuchungen und Behandlung!
PNH ist eine erworbene klonale Stammzellstörung, die durch die Produktion von abnormalen Erythrozyten, Granulozyten und Blutplättchen gekennzeichnet ist.
Der Defekt in den Erythrozyten macht sie anfälliger für komplementvermittelte intravaskuläre Hämolyse. Die Hämolyse führt zu einer Hämoglobinurie, die sich morgens als dunkel gefärbter Urin manifestiert. Der Begriff "Noctumal" bezieht sich auf die Überzeugung, dass die Hämolyse durch eine Azidose während des Schlafes ausgelöst wird, die die Komplementproteine zur Hämolyse der abnormalen Erythrozyten aktiviert; Dieses Konzept wurde jedoch widerlegt. Hämolyse tritt den ganzen Tag über auf, und tatsächlich ist die Hämolyse nicht paroxysmal; Der über Nacht konzentrierte Urin bewirkt jedoch eine drastische Farbänderung.
PNH besteht aus einer Triade von hämolytischer Anämie, Panzytopenie und Thrombose.
Die Patienten mit PNH können eine unterschiedliche klinische Darstellung haben:
1. Die PNH-Patienten können aufgrund der abnormen Anfälligkeit von Erythrozytenmembranen für die hämolytische Aktivität des Komplements eine hämolytische Anämie aufweisen.
2. Der PNH-Patient kann Thrombosen in den Gefäßen (z. B. hepatische, abdominale, zerebrale und unter dermale Gefäße) aufweisen.
3. Der PNH-Patient kann einen Mangel an Hämatopoese aufweisen, der leicht oder schwerwiegend sein kann, wie z. B. Panzytopenie bei aplastischer Anämie (PNH-Patienten haben eine 10 bis 20-prozentige Chance, eine aplastische Anämie zu entwickeln, wohingegen 5 Prozent der Patienten mit aplastischer Anämie leiden schließlich entwickeln sich PNH. Die Art der Verbindung zwischen diesen beiden Krankheiten ist nicht bekannt. Der Defekt in PNH scheint eine genetische Mutation zu sein, die dazu führt, dass der Anker für Glycosylphosphatidylinosit (GPI) nicht synthetisiert werden kann.
Der GPI-Anker bindet eine Reihe von Proteinmolekülen an die äußere Membranoberfläche, darunter die komplementregulierenden Oberflächenproteine [Decay-Activating Factor (DAF) oder CD55, homologer Restriktionsfaktor (HRF) oder C8-Bindungsprotein und Membraninhibitor der reaktiven Lyse (MIRL) oder CD59].
Diese Proteine auf der Zelloberfläche interagieren mit Komplementproteinen, insbesondere C3b und C4b, und dissoziieren die Konvertasekomplexe des klassischen und alternativen Pfads. somit werden die weitere Komplementaktivierung und die Auswirkungen der Komplementaktivierung (dh die Lyse von Zellen durch den Membranangriffskomplex C5b-C9) gestoppt.
DAF unterbricht normalerweise die Enzymkomplexe vom klassischen oder alternativen Weg, der C3 aktiviert. CD59 inhibiert die Umwandlung von C9 durch C5b-C8 in einen polymeren Komplex, der in der Lage ist, die Zellmembran zu durchdringen.
Abgesehen von den oben erwähnten komplementregulierenden Proteinen sind die folgenden Proteine auch auf den Zellmembranen von Patienten mit PNH vermindert oder fehlen.
ich. CD58 (Leukozytenfunktionsantigen 3)
ii. CD14 (Endotoxin-bindender Proteinrezeptor)
iii. CD24
iv. CD16a (Fey-Rezeptor).
PNH ist eine erworbene klonale Erkrankung aufgrund einer inaktivierenden somatischen Mutation eines Gens auf dem X-Chromosom in einer einzelnen abnormalen Stammzelle. Das für PNH verantwortliche Gen befindet sich auf dem X-Chromosom und wird als Phosphatidylinositolglycan A oder PIG-A bezeichnet. Mehr als 120 Mutationen des PIG-A-Gens wurden beschrieben.
Das betroffene Gen ist wichtig für die Biosynthese des GPI-Ankers. Die teilweise oder vollständige Abwesenheit des GPI-Ankers führt zu geringen Mengen oder Abwesenheit dieser Proteine an der äußeren Oberfläche der Zellmembran. Bis heute wurden auf der Oberfläche von Erythrozyten von Patienten mit PNH etwa 20 Proteine fehlen.
Bei einem Patienten mit PNH existieren sowohl normale Klone von Stammzellen als auch abnormale Klone von Stammzellen. Der Anteil der normalen und abnormalen Klonstammzellen variiert zwischen den Patienten und über einen bestimmten Zeitraum bei demselben Patienten.
Daher ist PNH derzeit als partieller oder vollständiger Verlust von GPI-Linked-Proteinen in einer Population von Zellen des hämatopoetischen Systems definiert. Abgesehen von Erythrozyten fehlen den Blutplättchen auch die Proteine auf ihrer Oberfläche. Die Lebensdauer von Blutplättchen wird jedoch nicht beeinflusst.
Das Fehlen von CD59 auf Blutplättchen führt zur Externalisierung von Phosphatidylserin, einer Stelle für Prothrombinase-Komplexe, und erhöht somit die Neigung zur Thrombose. Die Aktivierung des Komplements kann indirekt die Thrombozytenaggregation und Hyperkoagulierbarkeit stimulieren, was möglicherweise die in PNH beobachtete Neigung zur Thrombose erklärt.
WBCs haben neben RBCs und Blutplättchen auch diese Proteine auf ihrer Membranoberfläche.
Phänotypischer Mosaizismus in PNH:
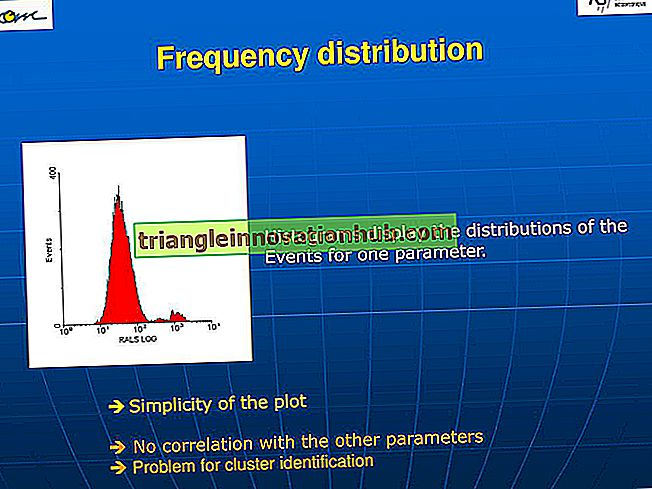
Anhand eines In-vitro-Tests zur Komplement-vermittelten Lyse von Erythrozyten, der die Sensitivität von Erythrozyten gegen Komplement quantifiziert, können drei Phänotypen von PNH-Erythrozyten identifiziert werden.
ich. PNH I zeichnet sich durch eine normale oder nahezu normale Sensitivität der Erythrozyten zum Komplement aus.
ii. PNH II ist von mittlerer Empfindlichkeit, etwa 3 bis 5 Mal anfälliger als normale Erythrozyten.
iii. PNH III zeichnet sich durch eine 15- bis 25-fache Empfindlichkeit gegenüber Komplement-vermittelter Lyse von Erythrozyten aus.
Bei Patienten mit PNH variiert der Anteil komplementempfindlicher und komplementunempfindlicher Zellen stark. Die Intensität der Hämolyse bei einem Patienten mit PNH hängt von der Größe der PNH-Population ab.
iv. Bei Patienten mit weniger als 20 Prozent der PNH III-Erythrozyten ist die Hämoglobulinämie mild oder nicht nachweisbar.
v. Mit einer 20 bis 50-prozentigen PNH III-Erythrozytenpopulation treten tendenziell schwere Hämoglobinurien auf.
vi. Konstante Hämoglobinurie ist mit> 50 Prozent der roten Blutkörperchen der PNH III-Region assoziiert.
PNH ist keine immunvermittelte hämolytische Anämie, die durch Autoantikörper entsteht. Vielmehr ist das Versagen, den alternativen Reaktionsweg der Komplementaktivierung auf Erythrozyten zu regulieren, für die Hämolyse in PNH verantwortlich. Der alternative Weg befindet sich in einem Zustand der kontinuierlichen Aktivierung.
Normale Erythrozyten haben Zelloberflächenkomplementregulationsmoleküle, die die Komplementaktivierung hemmen und Hämolyse verhindern. Im Gegensatz dazu fehlt den PNH-RBCs zwei wichtige Komplementmembranregulationsmoleküle (DAF und MIRL) und erliegen daher den während der Komplementaktivierung gebildeten Membranangriffskomplexen (C5b-C9), die zu Hämolyse führen. Die Halbwertzeit von komplementempfindlichen PNH-Erythrozyten beträgt nur etwa 6 Tage.
Von den zwei komplementregulierenden Proteinen ist MIRL wichtiger als DAF beim Schutz von RBCs vor komplementvermittelter Lyse. PNH ist eine seltene Krankheit. Beide Geschlechter sind gleichermaßen betroffen und es gibt kein familiäres Auftreten. Die Krankheit kann in jedem Alter auftreten und ist bei jungen Erwachsenen häufiger.
PNH ist eine chronische Krankheit mit einem mittleren Überleben von etwa 10, 3 Jahren. Die Morbidität hängt von der Schwere der verschiedenen Aspekte der Erkrankung, der Hämolyse, dem Knochenmarkversagen und der Thrombose ab. Die Haupttodesursache von Patienten mit PNH ist die Venenthrombose, gefolgt von Komplikationen des Knochenmarkversagens. Spontan-langfristige Remission oder Leukämie-Transformation des PNH-Klons wurde berichtet.
Klinische Merkmale:
Die Patienten mit PNH können bei jedem der drei Syndrome auftreten.
1. Patienten können mit intravaskulärer hämolytischer Anämie und dunklem cola-farbigem Urin auftreten. Die klassische Beschreibung von PNH ist der dunkle Urin in der Nacht mit teilweiser Lichtung während des Tages. Anfälle von Hämolyse können durch Infektion, Operation, Vollbluttransfusion, Injektion von Kontrastfarbstoffen oder sogar durch schweres Training ausgelöst werden.
2. Venenthrombose:
Lebervenenthrombose führt zu einem Budd-Chiari-Syndrom, das plötzliche und schwere Gelbsucht, Bauchschmerzen, eine schnell zunehmende Leber und eine Ansammlung von asketischer Flüssigkeit darstellt. Das Budd-Chiari-Syndrom kann zu Gefäßkollaps und Tod führen oder langsam und heimtückisch sein und zu Leberversagen führen.
Eine Abdominalvenenthrombose kann zu Bauchschmerzen führen und zu einem Darminfarkt führen. Milzvenenthrombose kann Splenomegalie verursachen. Hirnvenenthrombose: Sagitalvene wird häufig thrombosiert, was zu Papillenödem und Pseudotumor cerebri führt. Dermale Venenthrombosen können erhöhte, schmerzhafte und rote Knoten in der Haut verursachen; Die Knötchen können innerhalb von wenigen Wochen in der Regel ohne Nekrose abklingen.
3. Mangelhafte Hämatopoese führt zu Anämie. In einigen Fällen treten Neutropenie und Thrombozytopenie mit einem hypoplastischen Knochenmark auf, das einer aplastischen Anämie (aplastischen Episoden) ähnelt. Die aplastischen Perioden können Wochen bis Jahre dauern. PNH kann bei anderen Stammzellstörungen, einschließlich Myelofibrose, auftreten.
ich. Krampf der Speiseröhre kann am Morgen auftreten und klärt sich am Tag.
ii. Gleichzeitig mit der Hämoglobinurie kann bei Männern Impotenz auftreten.
Die Krankheit kann partielle Remissionen und Verschlimmerungen durchmachen.
Labor studien:
iii. Tests zur intravaskulären Hämolyse
ein. Serum-LDH ist erhöht.
b. Retikulozytenzahl wird erhöht
c. Fehlendes oder niedriges Serum-Haptoglobin.
d. Hämoglobinurie:
Die Hämolyse kann intermittierend sein und daher hängt das Testergebnis von der Zeit ab, zu der es durchgeführt wird.
iv. Hämosiderinurie ist fast ständig vorhanden und wird in der MRI- und CT-Untersuchung gesehen.
v. Direkter Coombs-Test ist oft negativ.
vi. Saccharosehämolyse-Test:
Saccharose liefert ein Medium mit geringer Ionenstärke, das die Bindung von Komplement an die roten Blutkörperchen fördert. Die gewaschenen Erythrozyten des Patienten werden mit ABO-kompatiblem normalem Serum und isotonischer (10%) Sucrose gemischt. Das Röhrchen wird 30 Minuten bei Raumtemperatur inkubiert, zentrifugiert und auf Hämolyse untersucht. (10% Hämolyse ist positiv und für PNH diagnostisch)
v. Der Zuckerwassertest wendet dasselbe Prinzip an, Blut mit Zucker zu mischen und auf Hämolyse zu prüfen.
vi. Acidified Serum-Test (Ham-Test): Im Acid-Serum-Test wird das Komplement durch den alternativen Weg aktiviert und führt zur Lyse abnormaler PNH-Zellen, die ungewöhnlich anfällig für Komplement sind. Die gewaschenen Erythrozyten des Patienten werden mit ABO-kompatiblem normalem Serum und Säure gemischt.
Nach einer einstündigen Inkubation bei 37 ° C werden die PNH-Zellen lysiert (In PNH werden normalerweise 10 bis 50% der Zellen lysiert.) Das eigene Serum des Patienten kann je nach verbleibendem Komplement im Serum zur Lyse führen. Ein positiver Test mit angesäuertem Serum tritt auch bei kongenitaler dyserythropoetischer Anämie Typ II auf (CDA-II oder erbliche Erythroblasten-Multikernität mit positivem Test mit angesäuertem Serum (HEMPAS)). In diesem Zustand tritt Lyse nicht mit dem eigenen Serum des Patienten auf. Diese Patienten haben einen negativen Zuckerwasser-Hämolysetest.
vii. Durchflusszytometrie unter Verwendung monoklonaler Antikörper gegen CD55 oder CD59 zum Nachweis von CD55 oder CD59 auf der Oberfläche von Erythrozyten und Granulozyten. Granulozyten bieten ausgezeichnete Ziele für die Durchflusszytometrie. Die Durchflusszytometrie zum Nachweis von CD55 und CD59 an Erythrozyten und Granulozyten ist dem Test mit angesäuertem Serum und dem Saccharose-Lyse-Test überlegen.
viii. Knochenmarkaspiration: Knochenmark ist normalerweise hyperzellulär mit normoblastischer Hyperplasie, kann jedoch hypozellulär sein. Bei einigen Patienten ist die aplastische Anämie die erste Diagnose und die Anzeichen einer PNH manifestieren sich später.
ix. PNH-Leukozyten haben einen niedrigen Leukozyten-Gehalt an alkalischer Phosphatase (der dem für chronisch myeloische Leukämie ähnlich ist).
x. Bildgebende Studien zur Untersuchung von Thrombosen.
Behandlung:
Die tägliche Verabreichung von Corticosteroiden während der Hämolyse und an anderen Tagen während der Remissionen verbessert die Hämoglobinwerte bei etwa 70 Prozent der Patienten mit PNH. Nährstoffe zur Verhinderung von Eisenmangel sind erforderlich; Eisenersatz kann jedoch die Retikulozytose stimulieren und neuere abnormale Erythrozyten freisetzen, was zu einer Verschlimmerung der Hämolyse führt. Die Zugabe von Prednison während der Eisenersatztherapie kann die Hämolyse verhindern. Tägliche Verabreichung von 1 mg Folsäure ist erforderlich.
Androgene Hormone stimulieren Erythropoisis und haben sich als nützlich erwiesen. Leuko-arme gepackte Erythrozyten minimieren die Alloimmunisierung. Gewaschene Erythrozyten verhindern eine Verschlimmerung der Hämolyse. Thrombotische Komplikationen erfordern eine Notfall-Heparin-Therapie, gefolgt von einem oralen Antikoagulans wie Coumadin. Manchmal kann Heparin das thrombotische Problem verschlimmern, wahrscheinlich durch Aktivierung des Komplements. Die Verabreichung von Cyclooxygenase-Inhibitoren wie Aspirin oder Ibuprofen verhindert dieses Problem.
Knochenmarkshypoplasie wird mit Anti-Thymocyten-Globulin (ATG) oder Knochenmarktransplantation behandelt. Wenn ein geeigneter Geschwisterspender verfügbar ist, sollte eine Knochenmarktransplantation so früh wie möglich in Betracht gezogen werden. Die üblichen Konditionierungsprogramme des Knochenmarkempfängers reichen aus, um die aberranten PNH-Klone auszurotten.